基因表达谱 的发展有助于科研工作人员进一步的理论知识充实及应用到研发等领域中。基因芯片是最近几年发展起来的基因表达重要工具,本文主要对这种技术的数据分析和管理方法作具体介绍。
一、引言
DNA微阵列(DNA microarray),也叫基因芯片,是近几年发展起来的一种能快速、高效检测DNA片段序列、基因型及其多态性或基因表达水平的新技术。它将几十个到上百万个不等的称之为探针的核苷酸序列固定在微小的(约1cm2)玻璃或硅片等固体基片或膜上,该固定有探阵的基片就称之为DNA微阵列。它利用核苷酸分子在形成双链时遵循碱基互补原则,可以检测出样本中与探阵阵列中互补的核苷酸片段,从而得到样本中关于基因结构和表达的信息。它的技术来源追溯到一个多世纪之前,Ed Southern发现被标记的核酸分子能够与另一被固化的核酸分子配对杂交 。
因此,Southern blot可被看做是最早的基因芯片。在八十年代,Bains W.等人就将短的DNA片断固定到支持物上,借助杂交方式进行序列测定。1995年,斯坦福大学开发出第一片cDNA芯片并用于生命科学研究,1998年美国Affymetrix公司将第一片带有13.5万个基因探阵的寡聚核苷酸芯片推向市场,标志着DNA微阵列的产业化,从此基因芯片或DNA微阵列的研究和应用得到了广泛的重视,可以说在生命科学研究界和产业界掀起了基因芯片热潮,1999年Nature出专刊介绍这门基因芯片及其应用。
基因芯片可用于DNA序列的再测序、基因SNP或多态性检测和基因表达分析。由于基因芯片技术是一种高通量检测技术,它可是并行的同时检测成百上千,甚至成千上万个基因的活动情况或DNA片段,改变了传统的每次只能检测一个基因的情况,因此能大大提高检测效率,降低检测成本,并保证了检测质量。
基因芯片技术可广泛应用于疾病诊断和治疗、药物筛选、农作物的优育优选、司法鉴定、食品卫生监督、环境检测、国防、航天等许多领域。它将为人类认识生命的起源、遗传 、发育与进化、为人类疾病的诊断、治疗和防治开辟全新的途径,为生物大分子的全新设计和药物开发中先导化合物的快速筛选和药物基因组学研究提供技术支撑平台。
通过基因表达谱的研究可以进行进一步的理论研究或应用研究。
1、理论研究。根据基因组基因表达谱可以进一步分析共表达基因是否存在共同的顺式调控元件,发现新的调控元件。此外,可以研究基因的调控规律,构建调控网络。
2、应用研究包括疾病诊断和药物开发。根据不同疾病状态下的差异表达谱的研究可以确定疾病的类型和进展。研究药物作用后基因表达谱的改变可以确定药物的毒性、预后和疗效,从而指导药物开发和临床合理用药。
在基于DNA微阵列的基因表达分析研究中,数据的分析和管理是一个关键性的问题,它直接影响了实验结果的准确型和实验的可靠性。

二、数据分析
数据的分析包括了三个部分:芯片图像处理获得单次实验的基因表达水平;整合多次实验得到基因表达矩阵;根据基因表达矩阵进行知识挖掘。下面简单介绍一下其中涉及的关键技术:包括归一化和聚类分析。
归一化对于cDNA微阵列技术,包含Cy3和Cy5两个通道,通常存在两个通道荧光亮度不平衡的问题,Cy3的亮度低于Cy5[Quackenbush, 2001]。归一化的目的是平衡实验过程中Cy3与Cy5两个通道的相对荧光亮度。
它基于如下的假设:芯片上的所有的基因,一组基因子集或一套外源的控制在标记前产生RNA,其平均表达率等于1。使用归一化因子调整数据,弥补实验的变化,“平衡”待比较的两个样本的荧光信号。主要有3种被广泛使用的技术用于来自同一个芯片杂交的基因表达数据的归一化。
1、总亮度归一化
总的亮度归一化数据依赖于假设:两个标记的样本的起始量是一样的,此外,假设一些基因在待检测的样本中相对于控制样本是上调的,另外一些是下调的。对于芯片上成百上千或成千上万的基因,这些变化应该是平衡的,因此,总的与芯片杂交的RNA的量是一样的。因此,芯片上所有的元素计算得到的总的累加亮度在Cy3和Cy5通道上是一样的,在这种假设下,计算归一化因子,并用于芯片上每个基因的亮度比例计算。
2、用回归技术归一化
对于起源于相关样本的mRNA,被分析的基因的显著性分数在相似的水平上被表达。在Cy5与Cy3亮度(或对数值)的散点图上,这些基因沿着直线聚类,如果两个样本标记和检测效率是一样的则该斜率将是1。这些数据的归一化等于用回归技术计算它的最合适斜率,调整各基因荧光亮度使计算得到的斜率为1。在许多实验中,亮度是非线性的,使用局部回归技术更合适,例如LOWESS(局部权值散点图平滑)回归。
3、使用比率统计归一化
Chen描述的基于比率统计的归一化方法。假设尽管在紧密相关的细胞中,单个基因可以上调或下调,RNA产生的总量与重要的基因近似相等,例如看家基因。基于这种假设,他们发展了一种近似概率密度比率Tk=Rk/Gk(R,G分别代表第k个元素的测量的红/绿亮度比)然后他们用于迭代过程,归一化平均表达率为1,计算可信度阈值用于识别差异表达的基因。
除了以上三种在应用中被广泛使用的除外,还有一些复杂的、非线性的方法用于归一化。归一化后,每个基因的数据以表达率或表达率的对数报告。应用对数值的优点是理解更简单,如果值大于0,则表示该基因的表达率大于1,反之小于1。
对于合成寡聚核苷酸微阵列不存在cDNA微阵列荧光不平衡导致的系统歪曲的问题,但是对于相比较的两组实验来说,需要用两块芯片与两个样本杂交两次,产生的原因包括两个样本中mRNA数量的差异或用于标记样本的染料的质量不同,都可能导致错误。在这里归一化的目的也是去除这些错误。
聚类分析
通过图1的数据获取过程,可以得到细胞的基因表达矩阵。基因的表达矢量定义为每个基因在表达空间的位置。用基因表达的观点看,每个实验在空间中表达一个隔离的和不同的轴,在该实验中的基因的测量值log2(比率)代表了几何坐标。
例如,如果我们有三个实验,对于一个给定的基因在实验1种的log2(比率)值是它的x坐标,在实验2中的值是y坐标,在实验3中的值是z轴,因此,我们能表示所有的信息,一个基因在x-y-z表达空间中用一个点表示。第2个基因,对于每个实验近似相同的值(log2(比率))将在表达空间中空间相近的点表示。不同表达模式的基因将于最初的基因离的较远。
对于更多的实验这种推广是直接的(尽管很难画出),表达空间的维度的增加与实验的数目相等。用这种方式,表达数据可以表示为n维表达空间,n是实验的数目,每个基因表达矢量表示为该空间内的单个点。
有了测量基因间距离的方法后,聚类算法根据在表达空间中的分离度选择基因和将基因分组。需要提及的是如果我们感兴趣聚类实验,我们将每个实验表示为一个实验矢量,包括每个基因的表达值。这里定义的实验空间,维度等于每个实验中分析的基因数目。同样的方法定义距离,我们能够应用任何的聚类方法来分析和分组实验。
为了解释多个实验分析的结果,直觉的可视化表示是很有帮助的。通常使用的方法依赖于表达矩阵的建立,矩阵的每一列表示单个实验,每一行表示特定基因的表达矢量。根据表达数据用不同的颜色表示矩阵元素建立多个实验的基因表达模式的可视化。表达矩阵有无数的方案来着色和表示。最常用的方法是根据每个实验的log2(比率)值,log2(比率)等于0用黑色,大于零的用红色表示,负数的用绿色表示。
对于矩阵中的每一个元素,相对亮度表示了相对表达水平,约亮的元素表示差异表达越大。对于任何特定的实验组,表达矩阵通常没有明显的模式或顺序。设计程序来聚类数据通常重组行、列或两者。当以这种方式可视表示可以看到明显的表达模式。
在聚类数据前,有两个问题需要考虑:
1、数据需要用某种调整方式来增强某一种关系?
2、采用何种距离测量来分组相关的基因。
在许多微阵列实验中,数据分析被具有最大数据值的变量决定,这样掩盖了其他重要的区别。为了避免这个问题,采用的一种方法是调整或重新确定数值范围,使每个基因的平均表达为0,称之为平均中心法过程。在这个过程中,基因的基本表达水平被每次实验测量值相减。这样增强了每个基因在每个实验中的表达水平的变异,而不考虑基因是否是上调或下调。这种方法对于分析时间过程的实验是特别有用的,可以发现在基础表达水平周围变异相似的基因。这些数据调整为-1~1之间的值。或者每个表达矢量的长度为1。
基因的聚类分析方法根据不同的描述包括:层次式与非层次式(k-means);分解法、合成法;有师(使用现有的生物学知识,关于功能相关的特定基因指导分类算法)、无师分析方法等。聚类分析技术非常有用,应该关注不同的算法、不同的归一化或者不同的距离矩阵,将把不同的目标放在不同的类中,此外,不相关数据的聚类仍将产生类,虽然他们没有生物学意义。因此基因表达分析方法的挑战是针对特定的数据应用适当的方法,使数据明显的分开。主要的无师聚类分析方法有层次式聚类法[Eisen,1998]、自组织神经网络[Tamayo,1999] 、k平均法、模糊聚类法等,有师分类包括矢量学习机法[Brown,2000]等,此外还有主元分析法和利用统计学的SAM法等。
1、层次式聚类法(hierarchical clustering method)
这是多元统计分析中常用聚类方法,对于n个样本构成的n个矢量,看作是n个类,先计算所有两类之间的相似性关系,将相似关系最近的两类生成一个新类,继续以上过程,直到最后只有一个类为止。在这个过程中每次形成一个新类,类的数目间减少一个,最后形成一棵树,反映样本之间的相似关系。
在计算新类与其它类的相似关系时有不同的方法,有最小距离法、最大距离法、平均距离法、重心法、离差平方和法等。如最小距离法是将组成新类的两个类分别与第三个类相似关系最近的值为新类和第三个类之间的相似关系。该类方法可以直观的反映基因之间的关系,而且计算速度快,但使用不同的类间距离计算法会产生不同的聚类结果,而且对于一个样本被分类后,就不能再参与分类,因此它不能将所有的数据作为一个整体进行分析,是一个局部决策的方法。同时当样本集非常大时,树型结果非常复杂,树的剪枝和类的确定比较模糊。
最简单、结果可视。是用于分析基因表达数据用得最多的方法,它是一种合成分析的方法,单个基因被连接形成组,继续直到形成单棵层次树。对于基因表达数据,平均连接聚类给出可接受的结果。主要问题是随着类数目的增加,表达某一类的表达矢量也许不再表示类种的任何成员。此外,与最初的基因顺序有关。
2、自组织神经网络法(SOM,self-organizing map)
自组织映射是Kohonen,T提出的类似大脑思维的一种人工神经网络方法,是一种竞争学习算法,可以被看作是一种将N维模式空间各点到输出空间少数点的映射。这一映射由系统本身完成,没有外部的监督,即聚类是以自组织的方式实现的。SOM采用无教师学习训练,训练完成后,分类信息存储在权值向量中,具有与权值向量相似的输入向量将分为一类。
包括1维和2维SOM,2维SOM也称为KFM(Kohonen Feature Mapping)。它们的区别在于KFM考虑邻近神经元的相互作用,即获胜神经元对周围神经元由于距离的不同会产生不同的影响。聚类结果与k平均法相仿,它的优点是自动提取样本数据中的信息,同时也是一种全局的决策方法,缺点在于必须实现设定类的数目与学习参数,而且学习时间较长。
3、模糊聚类法(Fuzzy Clustering method)
模糊聚类是模拟人类的思维方法,通过隶属度函数来反映某一对象属于某一类的不确定程度[15],从而建立起样本对于类别的不确定性的描述,准确反映样本之间的关系。模糊聚类分析方法的基本原理是将模糊数学中的有关概念与方法引进聚类分析,通过建立模糊相似关系来生成模糊等价关系,进而产生不同的水平截集,得到对样本的动态聚类结果。
由于动态聚类图的建立,可以方便的获取有明显特征的类,并能看到类的扩展,清楚地反映了类之间的关系,这样就克服了k平均法和自组织神经网络法必须事先确定类数目的缺点;同时对于每个λ值,所有的基因都重新参与分类,所以模糊聚类分析方法具有全局性,这是层次聚类法所不具有的。
4、k-平均法
它先将样本分成若干类,然后计算每类的中心矢量(每类样本的平均值),对于所有的样本重新计算与各类中心矢量的距离,然后根据距离调整分类,得到新的聚类中心,再次重复该过程,直到能满足一定条件为止。它是层次聚类法的很好的替代,其分类结果与SOM的聚类结果接近。主要问题是在聚类开始时必须指定类的数目。
5、主元分析法PCA(also called singular value decomposition)
主要思路是减少矢量的维数而不损失用于分类的信息。属于多元统计分析中一种常用的方法,它通过矩阵转换,有效的将对能对分类提供主要信息的参数提取出来,从而便于分析。
6、SVM(Support vector machine)方法
是机器学习的一种方法,它的最大的优点是用小样本可以将样本集分成若干类,但它需要一个学习的过程,通过学习确定核心机函数。
7、SAM(Significance Analysis of Microarrays)方法:
聚类分析虽然能发现一致的基因表达模式,但不能提供统计显著性的信息,用SAM方法来研究那一些基因会在用于癌症病人的致电离辐射疗法中产生副作用。这个问题是,每一次细微改变分析方法,得到不同的基因,使用一个非常低的辐射剂量,需要挑选出真正细小的变化。来自微阵列数据分析的最大的困难是确定哪一个结果是显著性的。SAM通过降低错误率和揭示哪一个基因被辐射影响解决了这个问题。
三、数据管理
DNA微阵列的应用,产生了大量的基因表达数据,现在有许多存储这些数据的数据库,通常与发表的论文结合起来,提供后来的研究者比较全面的信息。这些数据的共享、发布和再利用成为目前重要的研究内容。一些知名的研究机构如NCBI,EBI等正在试图建立新的标准,建立一些公共的知识库,如美国NCBI的Gene Expression Omnibus (GEO),英国EBI的ArrayExpress,日本DNA数据银行开发的基因表达库CIBEX。目前有一些比较有名的基因表达数据库:
ArrayExpress:
由EBI研究和开发。是基于基因表达数据的微阵列公共知识库。支持MGED(microarray gene expression data)组开发的MIAME(the minimum information about a microarray experiment)的各种技术指标。目的主要存储被很好注释的数据。ArrayExpress基于MAGE-OM对象模型,用Oracle实现,当前包含多个基因表达数据集和与实验相关的原始图像集。
ArrayExpress数据库接受MAGE-ML格式的数据递交或者通过MIAMExpress的基于Web的数据注释和递交工具。ArrayExpress提供一个简单的基于Web的数据查询界面,并直接与Expession Profiler数据分析工具相连,可以进行表达数据聚类和其它类型的直接通过Web的数据发掘。将进一步开发多个实验和数据库间的交叉查询。ArrayExpress数据库中的数据将与所有相关的由EBI维护的或再线的数据库相联接。
Gene Expression Omnibus
为了支持基因表达数据公共使用和分发,NCBI启动了GEO项目。GEO是一个基因表达和杂交阵列数据仓库,同时作为获取来自不同有机体的基因表达数据的在线资源。到2002年7月9日,数据仓库中包含内容:Platforms:99个(114M),
Samples 2170(1706M),Serials 61。Platform关于物理反应物的信息,平台类型如核酸、抗体和组织阵列和SAGE数据等的基因表达数据被接受、增加和归档作为公共数据集。Series是关于样本集的信息,样本间的相关和组织。
Stanford Microarray Database(SMD)
SMD存储微阵列实验的原始和归一化数据和对应的图像文件。另外,SMD提供数据获取、分析和可视化的界面。自从2002年1月1日,到6月3日,新增加789个新的阵列。达到总数2375个。45篇不同的论文。
3D-基因表达数据库(http://www.univie.ac.at/GeneEMAC/ )保存胚胎基因表达模式的三维模型和相关的使用GeneMAC方法根据系列组织学部分重建的形态学结构。
ArrayDB
软件包,提供交互式用户界面挖掘和分析微阵列基因表达数据,所有的分析表达数据来自微阵列实验。
BodyMap
人和老鼠基因的表达数据银行,在不同组织或细胞类型和不同时间。
Chip DB
可以根据基因分类、菌株、样本和实验查询。
ExpressDB
是关系型数据库包含酵母和大肠杆菌RNA表达数据,2000年10月,包含20m条来自众多出版物和内部研究的信息。
GXD(the gene expression database)
老鼠的基因表达数据
HuGE Index(Human Gene Expression Index)
目的是提供全面的数据库来理解人类基因在正常组织中的表达,现有19个组织59个样本的数据库
Yale Microarray Database (YMD)
多个实验室和研究中心的合作项目,包括微阵列图像的归档和通过查询语句查找,伴随着成百上千不同研究者的数据分析。
目前有几个因素阻碍了微阵列数据的广泛使用:
1、这是一个年轻的领域,仅仅是在最近才意识到需要识别数据的重要方面,以获取更多的信息。
2、基因表达数据比序列数据要复杂的多,仅仅在有具体的关于实验条件的描述时才是有意义的。与有机体的基因组相比,由细胞类型乘以环境条件一样多的转录本。
3、比较基因表达数据是相当困难的,因为目前,微阵列并不是在任何客观的个体上测量基因表达水平。事实上,大多数测量报告的仅仅是基因表达的相对变化,使用一个罕见标准化的参考样本。
4、不同的微阵列平台和实验设计以不同的格式和单位产生数据,用不同的方式归一化,所有这些使的这些数据的比较和集成是一种错误倾向的练习。
有许多实验室建立了自己的数据库,微阵列数据和论文用不同的格式在作者的网页站点上发布,目前大多数公共数据没有用足够的材料进行注释,供不同的独立小组使用。事实上,通常不进行注释。关于数据质量、可靠性和特定数据点可能的错误水平的所有细节被完全剥离了。例如,对于两通道的微阵列数据,通常仅仅给出信号去除背景后的比例,没有提示关于信号和背景水平的绝对信息,但是这些信息对于评价每一个基因表达的可靠性是很重要的。
有必要建立公共的微阵列数据仓库得到了公认。它的功能包括提供支撑基于微阵列实验的论文的数据的访问。这样的数据仓库在建设中,例如NCBI的GEO,日本的DNA数据库,和EBI开发的ArrayExpress,然而,那些必需的信息应该存在这些数据库中是不清晰的,存储原始的微阵列扫描图像,或每一个阵列元素最终的值(如两通道平台的每一个点的绿/红比率)是足够的吗?或者一些中间的数据,例如来自特定图像分析软件包的完整的输出?与原始数据发布或归一化的数据?实验中的那些信息是必须的?微阵列元素必须被注释使实验结果更容易被理解。
数据库中存储的信息必须有特定数据库或仓库的功能决定。如果仅仅是发表论文的数据支撑,对于实验的一些细节已经在论文中说明。建立论文与数据库的访问接口就行。这样的系统不大可能是有效的和可扩展的,更重要的是非标准化数据库的价值和使用是非常受限制的。例如,使用这些数据库对于高通量的自动化的数据分析和挖掘是非常困难的。过去几十年序列数据库的经历证明了在数据产生的早期阶段应用的结构和一致的注释的策略是很重要的。
对于微阵列实验相关的数据至少有三个层次:1、扫描图像(原始数据),2、图像分析过程得到的定量输出(微阵列定量矩阵);3、实验结果(基因表达数据矩阵)。来自微阵列研究的数据和注释必须满足一下要求:
1、关于实验的信息应该足够解释该实验,必须有足够详细的说明来与相类似的实验进行比较,允许实验的重复。
2、信息必须以某种方式结构化,保证有效的查询和自动化的数据分析和挖掘。
目前在基于微阵列的基因表达数据管理的主要成果是MIAME和MAGE-ML。
MIAME(the minimum information about a microarray experiment):
由微阵列注释工作组开发。目的是描述对于明白解释微阵列数据所必需的最少的信息,随后可以独立的验证这些数据。MIAME不是微阵列实验必须遵循的教条,而是一组指导方针,它将帮助微阵列数据库和数据分析工具的开发。MIAME中包含的信息如图2所示。
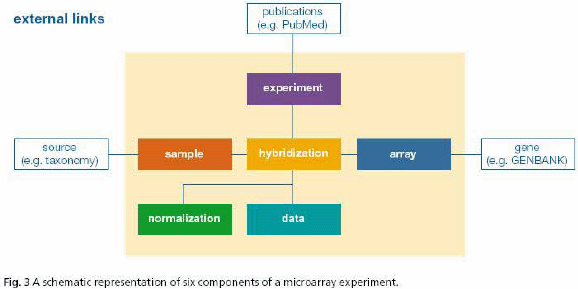
图2 MIAME的结构表示
MAGE-ML:
微阵列基因表达标记语言是一种语言,用来描述和基于实验的微阵列信息的通讯,它基于XML,可以描述微阵列设计、微阵列制造信息,微阵列实验组织和实施信息,基因表达数据和数据表达结果。MAGE-ML直接自动来自MAGE-OM, 后者是使用UML开发和描述——描述对象模型的标准语言。首先使用图形化表示法描述不同实体间的相互关系,比DTD更容易。然后,UML图表主要是针对人的,而DTD是面向计算机的。因此MAGE-OM可以认为是初级模型。
这两个标准已被许多大的基因芯片研究和制造机构采用,可以预言它们很可能将成为一种该领域的一个标准。
小结与展望
随着DNA微阵列技术的完善和在生命科学研究中的广泛应用,产生了大量的基因表达数据,这些数据中蕴含着大量的信息,如基因调控规律的信息,不同条件下表达差异的信息等等,利用这些信息可以进行基因启动子区域顺式调控元件的研究、基因表达调节途径或网络的研究、疾病或药物作用特异表达谱的研究等等。数据的增多直接带来的两个问题是数据的管理和知识发现。
数据的管理主要通过建立数据库的方式,目前已由较大的数据库服务器,这些数据间的共享和再利用迫切需要建立某种标准,从而提高利用效率,MIAME和MAGE-ML在这方面作了有益的尝试,有望成为一种规范。知识发现是从海量的数据中获取有生物学意义的信息,并形成新的生物学知识。在这方面的研究还处于初始阶段,最常采用的是统计学方法,如聚类分析、SAM等,但发展速度很快,目前已有大量的研究论文和分析软件。
目前,DNA微阵列尚属一个正在蓬勃发展中的年轻领域,这一方面虽然有不少科研工作成果,但总体上还远远不够,有不少因素阻碍了数据的分析和管理的发展,需要相关的工作人员进一步的努力,本文若有不足之处,还望指正。
科技日报北京4月1日电 (记者张梦然)美国马萨诸塞大学阿默斯特分校生物学家领导的跨学科小组最近发表了一项史无前例的研究:调查了18种灵长类动物的基因表达与大脑进化之间的联系。研究成果发表在《......
1月16日,中国科学院分子细胞科学卓越创新中心研究员周小龙、王恩多团队在《核酸研究》(NucleicAcidsResearch)上,发表了题为Multifacetedrolesoft6Abiogene......
DNA甲基化是最早发现的表观遗传标记之一,在真核细胞基因表达调控中发挥重要作用。随着DNA甲基化检测技术的进步,研究发现DNA甲基化具有完全甲基化和半甲基化两种状态,以及可以稳定遗传的半甲基化修饰。关......
细胞分裂素(cytokinin)是一种重要的植物激素,在植物的生长发育中扮演着多种角色,包括维持分生组织、促进维管组织分化、调控叶片衰老和促进再生等。以往研究表明,细胞分裂素的信号传递类似于细菌的双组......
在线质谱仪是一种用于工业生产流程中的质谱仪,主要用于工艺过程的在线气体分析及其他相关的检测。结构与功能:在线质谱仪通常包括进样系统、离子源、质量分析器、离子检测器、真空系统和数据处理系统。其中,进样系......
在线质谱仪的基本组成在线质谱仪的基本组成一般包括检测系统、真空系统、电控系统和数据处理系统几个部分。检测系统通常由进样系统、离子源、质量分析仪和离子检测器组成,参见图1。样品通过进样系统进入质量分析仪......
几十年来,科学家们已经知道,“垃圾DNA(junkDNA)”实际上起着至关重要的作用:尽管基因组中的蛋白编码基因提供了构建蛋白的蓝图,但是基因组中的一些非编码部分,包括以前被认为是“垃圾DNA”的基因......
12日,同时发表在《科学》《科学进展》和《科学·转化医学》杂志上的21项研究,公布并详细解释了迄今为止最全面的人类脑细胞图谱。这些研究对3000多种人类脑细胞类型进行了特征分析,阐明了某些人类脑细胞与......
8月7日,中国科学院动物研究所翟巍巍/马亮团队在《自然-通讯》(NatureCommunications)上,发表了题为SONARenablescelltypedeconvolutionwithspa......
急性肾损伤(AKI)的发病率和死亡率很高。肾损伤分子-1(KIM1)在肾小管损伤后显着上调,并作为各种肾脏疾病的生物标志物。然而,KIM1在AKI进展中的确切作用和潜在机制仍然难以捉摸。2023年7月......