10年时间,花费10亿美元,研发一款新药,无论是利益的驱动,还是拯救万千患者的成就感,药企的这一行为都值得我们尊敬。
一款新药从研发到上市都需要经过哪些流程?每一步又有哪些经验可以借鉴?本文以小分子药物为例,试着做了一个梳理,希望能对您有所帮助。
1.研究开发(一般 2-3年)
实验室研究,寻找治疗特定疾病的具有潜力的新化合物。
1 、药物靶点的发现及确认
这是所有工作的起点,只有确定了靶点,后续所有的工作才有展开的依据。
2 、化合物的筛选与合成
根据靶点的空间结构,从虚拟化合物库中筛选一系列可匹配的分子结构,合成这些化合物,它们被称为先导化合物。
3 、活性化合物的验证与优化
不是所有先导化合物都能符合要求,在这个阶段需要通过体外细胞试验验证,初步筛选出活性高、毒性低的化合物,并根据构效关系进行结构优化,这些化合物称为药物候选物。
同时也存在一个化合物对目标 A 靶点没有作用,却有可能对其他的 B 靶点、C 靶点有非常好活性的情况,暂且不表。
2.临床前实验(一般 2-4年)
这一阶段目的,一是评估药物的药理和毒理作用,药物的吸收、分布、代谢和排泄情况(ADME)。二是进行生产工艺、质量控制、稳定性等研究(CMC)。
第一部分的实验需要在动物层面展开,细胞实验的结果和活体动物实验的结果有时候会有很大的差异。这一步的目的是确定药物的有效性与安全性。
第二部分需要在符合GMP要求的车间完成。
1 、药理学研究
包括:药效学、药动学
2 、毒理学研究
急毒、长毒、生殖毒性,致癌、致畸、致突变情况
3 、制剂的开发
总不能弄点化合物就直接往嘴里倒吧,制剂开发是药物应用的一个重要环节。比如有的药口服吸收很差,就需要开发为注射剂。
有的药在胃酸里面会失去活性,就需要开发为肠溶制剂。
有的化合物溶解性不好,这也可以通过制剂来部分解决这个问题。
还有的需要局部给药,就需要通过制剂开发成雾化剂、膏剂等。
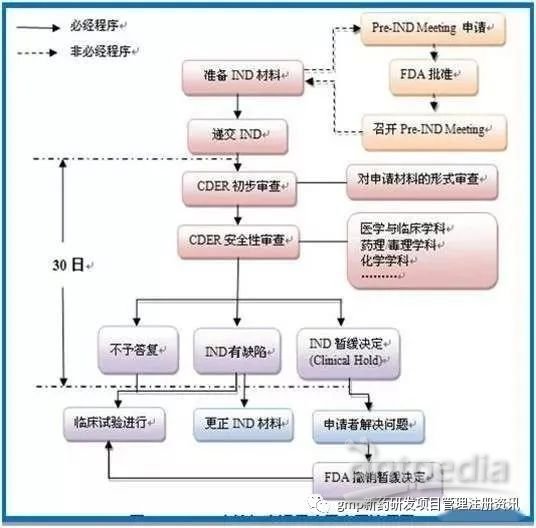
• Ⅰ期临床 20-100例,正常人,主要进行安全性评价。 • Ⅱ期临床 100-300例,病人,主要进行有效性评价 。 • Ⅲ期临床 300-5000例,病人,扩大样本量,进一步评价。 因为Ⅰ-Ⅲ 期临床在整个药物研发过程中非常重要,我们重点讲一下这部分。 传统意义上,新药的临床研究分为I/II/III期,后来 II 期又分成 IIa 和 IIb(很大程度上是因为肿瘤药物研究),接着出现了 0 期研究的概念(很大程度上也是因为肿瘤药物的研发)。然后又有人提出,0/I 期为早期临床研究,IIa 为中期临床研究,IIb 和 III 期为晚期临床研究。 这种分类,其实我觉得在两个方面对我们进行临床研究设计的人来说,是很重要的: 1. 分期是和研究目的相关的,不同的目的,可以分到不同时期/阶段的。 2. 不同时期/阶段的临床研究,往往对应于一定的研究框架设计(交叉研究,平行对照,单臂,终点选择等等)。 所以,理解不同时期/阶段的临床研究,其意义已经超出了知道有这样的分法本身。 分期的一头是研究目的,另一头是对应的设计框架(当然也只是粗略的框架,很多问题,要根据疾病、药物等来确定),理解分期,有助于我们了解,一个研究目的,大体上有哪些设计方法和思路,大体上可以如何去处理;为什么该如此进行,各种设计的优缺点在哪里。 所以,我们先看一下不同阶段的研究目的。 1.0 期 目的是在满足一定的统计学要求的前提下,在有限的样本量(通常不超过20)、有限的时间内(每个患者的治疗期常不超过数周),初步判断一个研究药物是否有效、某个剂量是否有效,是否应继续开发下去,附带看一下某些疗效判断方法是否可行。 从目的角度,0 期研究也是初步判断药物的效果,类似 II 期(尤其是IIa),所以 0 期的设计,在某些方面类似 IIa 的单臂研究,但是由于样本量小、又要满足一定的统计学要求,所以也有其特点,设计起来其实可能更为复杂。 2.I 期 通常是摸索剂量(dose finding, dose-ranging),药代(PK)和药动(PD),样本量也不大(一般不超过20,或者20左右)。尤其是PD,通常会使用交叉研究,因为交叉研究有其特点,适合此类设计目的。 交叉研究中,每个患者都是其自己的对照(在不同阶段,分别扮演研究组和对照组的角色),而且交叉研究的变量一般小于平行对照,所以在利用样本方面,交叉研究的效率远高于平行对照,交叉研究如果纳入 20 例患者,平行对照 50 例都不一定能获得相似的结果。 但是交叉对照也有很多限制,例如脱落和失访对交叉研究的影响远大于平行对照研究;同一个人,不同时期接受不同治疗、中间隔以洗脱期,导致后遗效应,以及治疗-时间相互作用等等。所以在后期临床研发中,很少看到有用交叉对照的(有,但远少于平行对照)。 3.II 期 II 期是初步判断疗效的和安全性的(其实安全性贯穿研发始终),所以一般称为safety & activity研究(SA研究)。有多种做法,根据不同的临床疾病环境,以及既往数据等,可以采取单臂,平行对照的 RCT 等,终点一般选择 surrogate,而不是 clinical outcome。如果分 IIa 和 IIb,则 IIa 往往是单臂,样本量一般不会过百;IIb 则往往是平行 RCT。 4.III 期 III 期是严格的验证药物效果的验证性临床研究。很多会议上介绍临床研究,往往以此类研究为模版进行介绍,在假设检验的框架下进行介绍。
NDA申报资料 — CTD(Common Technical Document)
CTD主要由五大模块组成: 流程: ①批准信 符合要求,可以上市 ②可批准信 ③拒绝信 ●NDA特殊审评程序 ①优先审评(Priority Reviews) ②加速审批(Accelerated Approval) ③快速通道(Fast-track) 以万络为例,1998年11月23日万络提交NDA 申请,编号 21-042,“1999 年 5 月 20 日获 FDA 批准,历时 178 天。 临床监测期:IV期临床 受试者要大于 2000 例,同时要进行社会性考察。 仍以万络为例:2000 年进行了“VIGOR”胃肠道试验 ——显示较少的胃肠道副作用, 但是使用 18 个月后会引发 2 倍的心脏病/中风风险。 2001 年,“APPROVe”腺瘤息肉预防试验 ——服药超过 18 个月出现较高的心血管疾病风险。 目的:重新审核 NDA 中的有效性和安全性。 万络,2002 年 4 月:默克公司增加了万络可能出现心血管副作用的警告。 2004 年 9 月 28 日,默克公司与 FDA 商讨有关万络实验结果的事宜。 2004 年 9 月 30 日, 再审评:“万络”,由默克公司主动召回。
①行政和法规信息
②概述:药物质量、非临床、临床试验的高度概括
③药品质量详述
④非临床研究报告
⑤临床研究报告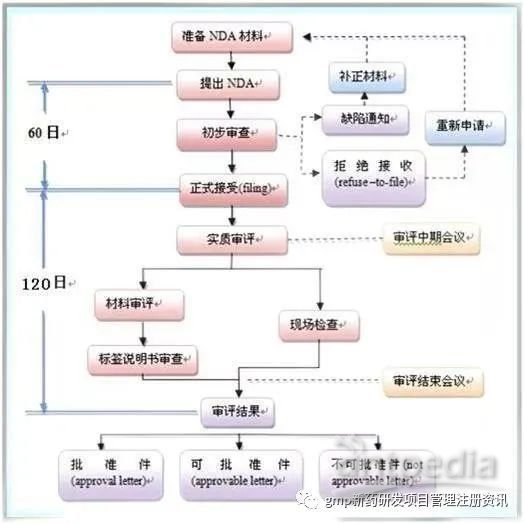
基本满足要求,少数不足可以修改 。
申请人应在收到10日内作出回应修正,否则视为自动撤回。
存在严重问题或需要补充大量信息资料。
申请人可在10日内提出修正或在30日内要求听证
适用于能够在治疗、诊断或预防疾病上比已上市药品有显著改进的药品,优先安排NDA审评。
用于治疗严重或危及生命疾病的药品,且存在合理并能够测量的“替代终点”(Surrogate endpoint),即药物预期的治疗效果的指标,变通审评标准,利用“替代终点”审评。
用于治疗严重或危及生命疾病的药品,且有潜力满足临床尚未满足的医学需求,早期介入,密切交流,分阶段提交申报资料。5、上市后研究
6、上市后再审批(一般上市后4-10年)
2025年6月,国家药监局共批准注册医疗器械产品225个。其中,境内第三类医疗器械产品184个,进口第三类医疗器械产品26个,进口第二类医疗器械产品14个,港澳台医疗器械产品1个,包含体外诊断产品50......
十年积淀,第十届中国先进诊断技术开发与应用论坛将于2025年8月14-15日在杭州举办,聚焦诊断技术在感染与病原微生物、AD等领域的应用,AI/tNGS/mNGS/多重PCR/核酸质谱等新技术的创新;......
IVD在2025年全行业的困局,在2024年最后一天的那场史上最大规模集采已露端倪。但2025年一季报的集体披露,让严酷的生存挑战呈现得更为清晰。从数据来看,头部外资IVD企业正面经受了第一波冲击波。......
2025年4月17日-19日,中国分析测试协会标记免疫分析专业委员会2025年学术峰会于江西九江盛大召开。本次会议由中国分析测试协会标记免疫分析专业委员会主办,九江学院、九江市医学会检验医学分会协办,......
2025年4月17日-19日,由中国分析测试协会标记免疫分析专业委员会举办的“中国分析测试协会标记免疫分析专业委员会2025年学术峰会”在江西九江盛大召开。本次大会以“促进精准诊断、维护健康生活”为主......
近日,帝肯从Revvity的子公司CisbioBioassays收购ELISA免疫测定资产。 TecanGroup周四宣布,将从Revvity子公司CisbioBioassays收购与ELI......
各大模型开发企业,各有关单位:为深入贯彻落实自治区党委、政府关于“人工智能+”行动的决策部署,积极推进人工智能赋能千行百业发展,广西科技厅聚焦特色优势产业,围绕行业龙头企业亟待解决的关键技术难题,遴选......
3月6日,由中国科学院上海药物研究所(以下简称上海药物所)、浙江大学、烟台新药创制山东省实验室和中科中山药物创新研究院联合申报的化药1类新药TLX-83胶囊,成功获得国家药品监督管理局颁发的临床试验通......
2025年3月10日,国家药品监督管理局(NMPA)发布了最新一批药品批准证明文件送达信息。此次共有133个受理号获批,涵盖多款重磅新药,涉及辉瑞、艾伯维、阿斯利康、科伦博泰等知名药企。在本次获批的药......
全国政协委员、中国医学科学院肿瘤医院肝胆外科副主任赵宏4日在全国政协十四届三次会议首场“委员通道”上表示,过去一年,我国批准上市创新药48个、创新医疗器械65个。我国在研新药数量已跃居全球第二位。更多......